
SARS-CoV-2: what's in a name? Everything.
Evidence suggests that certain parties were pushing hard for a name that suggested novelty.


“Naming is everything” might be a trite observation, but that doesn’t make it any less true.
Sigmund Freud’s nephew Edward Bernays, popularly regarded as the father of the discipline of public relations1, used to advise charities for free, and one such piece of advice was to rename multiple sclerosis as “MS” as the longer form was too difficult for the public to remember2.
This article offers a basic primer on the naming of new products. It suggests that choosing the correct name can offer these benefits.

I have found evidence that certain actors in the pandemic narrative attached some importance to naming the pathogen now known as SARS-CoV-2. Perhaps they were particularly cogniscent of this, taken from the above:
Visibility helps a product stand out in a competitive marketplace against similar products.
This paper on the naming of the "new virus" was published as preprint on 11 Feb 2020. It a statement of the Coronavirus Study Group, part of the International Committee on Taxonomy of Viruses (ICTV)3.
It makes interesting reading. Notably, it contains the following:
Likewise, we know that RNA viruses persist as a swarm of co-evolving closely related entities (variants of a defined sequence, haplotypes), known as quasispecies.
Their genome sequence is a consensus snapshot of a constantly evolving cooperative population in vivo and may vary within a single infected person and over time in an outbreak.
If the strict match criterion of novelty was to be applied to RNA viruses, it would have qualified every virus with a sequenced genome as a novel virus...
This is entirely consistent with what those of us who have been arguing all along, ie:
There was nothing novel around
All was found - by shining the spotlight of PCR on it - was part of the endemic and constantly mutating coronavirus swarm
In the paper, they report on the genome of the “new” virus, discuss its variation from known coronaviruses, and conclude thus:
The above results show that, in terms of taxonomy, SARS-CoV-2 is (just) another virus in the species Severe acute respiratory syndrome-related coronavirus. In this respect, the discovery of this virus differs considerably from the description of the two other zoonotic coronaviruses, SARS-CoV and MERS-CoV, introduced to humans in the 21st century (Fig. 5A). Both these viruses were considered novel by this study group based on prototyping two species"
This is basically the group stating that they thought that SARS1 was novel, but not SARS-CoV-2!
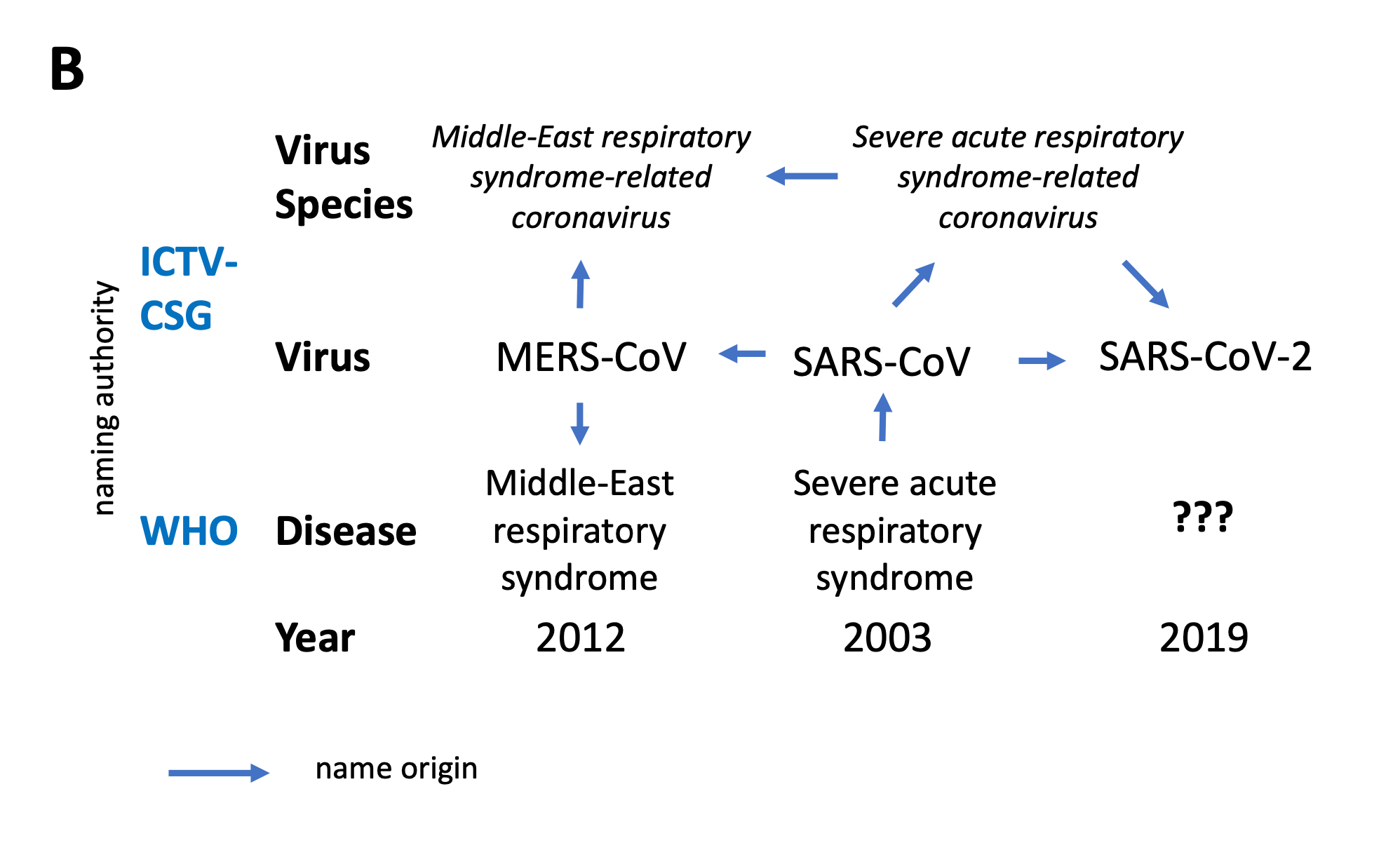
“Fig 5A” in the above quote refers to the first of 2 panels which the paper’s authors describe as: “Virus novelty and naming of the three zoonotic coronaviruses emerging in the first decades of the 21st century. Year, indicates the year in which the virus was first identified.”
The first panel is labelled:
Independent assessments of virus novelty by the ICTV-CSG and WHO performed during the three outbreaks came to different conclusions. Vertical arrows indicate the degree of virus novelty according to taxonomy.

The 2nd panel illustrates the History of coronavirus naming during the three zoonotic outbreaks in relation to virus taxonomy and disease (clinical manifestation).
In another part of the paper the authors write:
Although SARS-CoV-2 is NOT a descendent of SARS-CoV4 and the introduction of each of these viruses into humans was likely facilitated by unknown external factors, the two viruses are genetically so close to each other that their evolutionary histories and characteristics are mutually informative. Our understanding of these pathogens could be significantly advanced if both viruses were characterized along with viruses of other origins, known and yet-to-be discovered, as part of the Severe acute respiratory syndrome-related coronavirus species, with the long-term goal of comprehending the biology and evolution of that species, as is the norm elsewhere in biology. To connect this development to health care, diagnostic tools that target the entire species should complement existing tools that detect individual pathogenic variants.
In other words the paper itself suggested that the response and the research should have been based on what was assumed about SARS1 (the disease said to have been caused by SARS-CoV), not starting from scratch as a novel virus.
So, as at the publication date of this pre-print:
WHO were pushing for SARS-CoV-2 to be regarded as novel but the committee tasked with examining features of new viruses and giving them a name DID NOT agree.
Panel B isn’t 100% clear, but it suggests that WHO don’t seem at all sure about how this infection manifests clinically. Now I suppose it is possible to say that the question marks only suggest that they aren’t sure how to name the disease, but this quote from the paper does not support that interpretation at all:
The spectrum of clinical manifestations associated with SARS-CoV-2 infections in humans remains to be determined.
This seems odd given that as of the date of this publication, the Chinese had:
Already reported its cluster of atypical pneumonias in Wuhan
Determined the sequence of “the virus” from their first patient, a man with an unremarkable respiratory infection
Reported well over ten thousand cases
All that without being sure what the symptoms of this novel disease really were?
Later in Nature, this final “peer-reviewed” version appeared - now described as a “Consensus Statement”5.
This article was actually initially submitted on 5 Feb 2020, BEFORE the pre-print above was published, and accepted for publication on 19 Feb 2020.

Comparing this version with the pre-print is informative.
Nearly the entirety of the above quotes from the pre-print has been removed - including the 2 panels illustrating the assessment of novelty / clinical manifestation.
There is no mention of the “swarm of co-evolving closely related entities (variants of a defined sequence, haplotypes), known as quasispecies”
The phrase “in terms of taxonomy, SARS-CoV-2 is (just) another virus in the species Severe acute respiratory syndrome-related coronavirus...” has also disappeared.
Other doubts about whether SARs-CoV-2 was a truly novel pathogen causing a novel disease seem to have been deleted or watered-down. Although this part is preserved:
Although SARS-CoV-2 is NOT a descendent of SARS-CoV and the introduction of each of these viruses into humans was likely facilitated by unknown external factors, the two viruses are genetically so close to each other that their evolutionary histories and characteristics are mutually informative.
…this, which followed it in the pre-print has gone:
Our understanding of these pathogens could be significantly advanced if both viruses were characterized along with viruses of other origins, known and yet-to-be discovered, as part of the Severe acute respiratory syndrome-related coronavirus species, with the long-term goal of comprehending the biology and evolution of that species, as is the norm elsewhere in biology.
In its place is a paragraph which makes a rather poor attempt at hiding its key message - “please send for us to spend on our shiny new science” - in a sea of word salad mumbo-jumbo (my emphasis below):
The currently known viruses of the species Severe acute respiratory syndrome-related coronavirus may be as (poorly) representative for this particular species as the few individuals that we selected to represent H. sapiens in Fig. 1. It is thus reasonable to assume that this biased knowledge of the natural diversity of the species Severe acute respiratory syndrome-related coronavirus limits our current understanding of fundamental aspects of the biology of this species and, as a consequence, our abilities to control zoonotic spillovers to humans. Future studies aimed at understanding the ecology of these viruses and advancing the accuracy and resolution of evolutionary analyses41 would benefit greatly from adjusting our research and sampling strategies. This needs to include an expansion of our current research focus on human pathogens and their adaptation to specific hosts to other viruses in this species. To illustrate the great potential of species-wide studies, it may again be instructive to draw a parallel to H. sapiens, and specifically to the impressive advancements in personalized medicine in recent years. Results of extensive genetic analyses of large numbers of individuals representing diverse populations from all continents have been translated into clinical applications and greatly contribute to optimizing patient-specific diagnostics and therapy. They were instrumental in identifying reliable predictive markers for specific diseases as well as genomic sites that are under selection. It thus seems reasonable to expect that genome-based analyses with a comparable species coverage will be similarly insightful for coronaviruses. Also, additional diagnostic tools that target the entire species should be developed to complement existing tools optimized to detect individual pathogenic variants (a proactive approach). Technical solutions to this problem are already available; for example, in the context of multiplex PCR-based assays42. The costs for developing and applying (combined or separate) species- and virus-specific diagnostic tests in specific clinical and/or epidemiological settings may help to better appreciate the biological diversity and zoonotic potential of specific virus species and their members. Also, the further reduction of time required to identify the causative agents of novel virus infections will contribute to limiting the enormous social and economic consequences of large outbreaks. To advance such studies, innovative fundraising approaches may be required.
Concluding remarks
All of the above strongly suggest that there was - initially at least - no real agreement in respect of whether a truly novel pathogen had actually been identified. However, revisions to the manuscript turned a nuanced statement into a “Consensus Statement” which appears to argue strongly in favour of novelty, removing or watering down the elements which introduced any doubts as to this conclusion.
What science changed in those few short weeks?
AKA propaganda.
They don’t really tell us how they could be so sure of that.
Remember: if you don’t agree with “consensus” you are “anti-science”.
Tedros of the WHO renaming monkey pox as M-Pox, might have been trying to give the condition a 'higher profile', then he declared it to be a PHEIC.
We already know that Fauci in the autumn of 2019 is on record in a meeting saying that people weren't sufficiently scared of the term 'flu. A very revealing assertion. What arrived a few short months later but the blaze of propaganda for SARS-COV-2/ Covid-19.....New terms to describe a flu like illness, made 'scary'....'anyone could get it'! No one was safe unless they obeyed instructions to stay indoors, not go to school, submit to a 'vaccine'. How lives were ruined, society destabilised, economies degraded ....all down to new labels for illness which appears seasonally across the globe, and generally affecting the old, infirm, those with underlying morbidity. Beware then a newly named / rebranded illness. Swine Flu mark 2? No...something with more impact...'Arcane Avian flu'...the double A brand...not to be ignored but understood only by the select few!
Don't you think it's worth mentioning that the authors included Baric and Drosten - both people with an interest in bolstering the zoonosis hypothesis? What changed was presumably the reviewers insisting that they tone down their exaggeration that it was all natural.